GAA (Glucosidase Alpha Acid)[]
GAA is a gene that encodes for glucosidase alpha acid, an enzyme that contributes to glycogen metabolism in vertebrates. Glucosidase alpha acid hydrolyzes glycogen, a complex carbohydrate, to form glucose, a sugar monomer, in the lysosome of the cell. The enzyme will cleave the a-1,4 and a-1,6 linkages in glycogen, maltose, and isomaltose in the lysosome. Without GAA glycogen accumulates within various organelles.
Other names for glucosidase alpha acid include acid alpha glucosidase, lysosomal alpha-glucosidase, acid maltase, alpha 1,4 glucosidase, amyloglucosidase, and glucoamylase.
The gene is located on the long arm of chromosome 17, between the bases 78,075,354 and 78,093,678. Chromosome 17 is shown below and GAA is located in the q25.3 region.

{kind=link}
GAA location on chromosome 17 is in region q 25.3. http://useast.ensembl.org/Homo_sapiens/Location/View?db=core;g=ENSG00000171298;r=17:78075355-78093678;t=ENST00000390015

{kind=link}
Graphical view of GAA gene. Entire GAA sequence is included as well as mRNA, exons, and CDS. Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A (2012) Geneious v5.6, Available from http://www.geneious.com
The actual GAA gene is shown to the left. This gene contains 20 exons, however the first amino acid is encoded in exon 2 and the last amino acid is encoded in exon 20.
GAA is also found in mice (mus musculus), the brown rat (Rattus norvegicus), cattle (Bos taurus), and the chimpanzee (Pan troglodytes).
Glycogen Metabolism[]
Glycogen is a glucose polymer that is degraded in energy deficient states to glucose, the body's primary source of fuel/energy. Glycogen stores exist within a variety of tissues, but are most notable in the liver and modestly present in muscle. These macromolecule resevoirs reside in large cytosolic granules with each glycogen strand composed of about 55,000 glucose molecules. During exercise, in-between meals, and during fasts glycogen stores are depleted in both the muscle and the liver. The glycogen may be degraded in one of two ways. One option is through classic glycogenolysis where the breakdown of this macromolecule is through glycogen phosphorylase and debranching enzyme AGL. This results in free glucose-1-phosphate from cleavage of a-1,4 linkages and free glucose molecules from the cleavage of a-1,6 linkages. In the other pathway, glycogen is transferred to the lysosome and hydrolyzed into glucose molecules by GAA. Unfortunately, this second pathway is poorly understood.
Interestingly, 10% of the liver's glycogen is in the lysosome. Currently, there are several ideas regarding the presence of glycogen within the lysosome. It is thought that glycogen degradation in the lysosome is related to autophagy of abnormally phosphorylated and branched glycogen. It is also thought that "glycogen autophagy" is of importance in newborns. Glycogen stores are present in the liver and heart of newborns. As gluconeogenesis is not fully developed at birth, lysosomal degradation of these stores supplies the infant with energy.
Pompe Disease[]
Pompe disease, also known as glycogen storage disease

{kind=link}
Pompe Disease in Mouse Skeletal Muscle. 3 Dark ovals are lysosomes filled with glycogen. http://www.erasmusmc.nl/klinische_genetica/research/pompe_center/ziekte/?lang=en
II (GSD II), is an autosomal recessive disease in which the GAA gene is mutated in both copies. When this occurs individuals are unable to breakdown glycogen and so glycogen accumulates within the cells. Glycogen build up can then reach toxic levels causing organ damage and the often associated muscle damage. Toxicity and damaged have been attributed to glycogen displacing other organelles within the cell and through the lysis of the lysosome causing cell death.
More than 200 mutations have been identified within the GAA gene contributing to the development of Pompe disease. These mutations include missense, nonsense, and frameshift mutations where the change of one or more amino acids induces the disease.
The disease is classified and will present itself during several life stages: infancy, childhood, or adulthood designated infantile onset, non-classic infantile onset, and late onset respectively. The estimated frequency of the disease is 1 in 14,000-250,000.
Classic Infantile Onset[]
Several months following birth infants may be diagnosed with the classic form of the disease. Infants often have poor muscle tone, weak muscles, slower growth, breathing problems, enlarged liver, and heart defects. The long-term prognosis is not favorable as many infants will die in childhood.
Non-Classic Infantile Onset[]
This form of the disease is usually observed after the age of one. Similar to classic onset, children will have progressive muscle weakness, poor muscle tone, and difficulty breathing. Children will also have delayed motor skills. Death in childhood is common with this form of the disease.
Late Onset[]
The late onset form may appear in childhood or adulthood. This form of the disease is thought to be milder although patients also suffer from progressively weak muscles and potentially breathing problems. The heart is not effected in the same manner.
Genotype-Phenotype Correlation[]
As previously mentioned a large number of mutations have been associated with GAA (over 200). Several studies have looked at observed mutations in the GAA gene (related and not related to Pompe disease). One group of studies have evaluated the frequencies of several mutations found in different populations and their effects.
- One of these mutation was in African-Americans (c.2560C>T) and could be traced back to a small village in North Africa. This mutation produces a premature stop codon, generating in a fragment of the GAA protein. Apparently, this mutation spread along the West African Coast to Nambia. The same mutation was also found in a Saudi Arabian family.
- Another mutation found (c.1935C>A) induces a change in amino acids at position 645 from Asp to Glu. This mutation is present in high frequencies in Taiwan and the Chinese coast.
- Asian populations also had higher frequencies of other mutations (c.1726G>A, c.2065G>A). c.1726G>A reduces GAA levels and activity. c.2065G>A changes the net charge of the protein, with limited effect . These mutations are often observed together and approximately 3.3-3.9% of the Asian population are homozygous for these mutations. While they have very low levels of GAA, they do not present with the Pompe phenotype.
- A common GAA mutation has been observed in Caucasian populations (c.2481+102_2646 +31del)
- In Caucasian children the mutation c.-32-13T>G is observed in the first intron of GAA. This mutation leads to improper splicing of the pre-mRNA in 80-90% of pre-mRNA. This mutation is also thought to have been passed along through the generations.

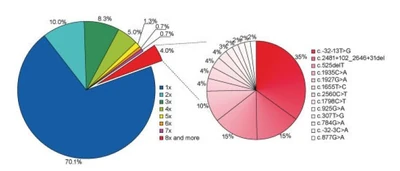
{kind=link}
Diversity of GAA mutations. Percentage of mutations uniquely observed and mutations collectively observed in 393 sequences. "The genotype-phenotype correlation in pompe disease)
Despite these findings, further evaluation has shown that most mutations occurring in the GAA gene are unique and rare. Given 393 mutated sequences of the GAA gene (with 257 defined as pathogenic), 70% were only seen in one individual. (see figure to left).

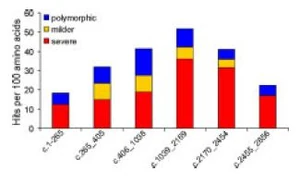
{kind=link}
Distribution of various observed GAA mutations in the GAA gene. "The genotype-phenotype correlation in pompe disease"
The aforementioned mutations are distributed throughout the GAA gene. The figure to the left demonstrates this distribution and the changes these mutations induce in the protein (i.e. severe would be a drastic change in the protein that effects its function).
Given the variability in the mutations, researchers have not been able to link specific mutations with different forms of Pompe disease. Studies have attempted to predict the effect of various sequence mutations on the Pompe phenotype using algorithms, but have not yielded reliable results. However, the Pompe phenotype is linked with enzyme activity. Individuals with severe mutations present with Classic Infantile Onset and have very very little (if any) GAA activity at birth. When considering late onset in the adult population, the inverse is true. These individuals have milder mutations, present with low levels of GAA and low activity levels of GAA their entire life. Therefore GAA expression and activity is linked to different disease forms.
As a relevant aside note: while Pompe disease is classified as an autosomal recessive disease, individuals with one mutation often present with less GAA activity (up to a 50% loss). However, these individuals are not considered to have the disease. A standard for Pompe diagnosis often includes having a 2.5 times less GAA activity than expected.
Treatment[]
Currently, enzyme replacement therapy is being utilized. This however is not without problems. Patients may develop an immune response to the recombinant human enzyme administered. This response is most prevalent within the classic infantile onset. To increase survival rates, pateints presenting with classic infantile onset may also be given anti rhGAA IgG antibody titers in concert with enzyme replacement therapy. (Currently Myozyme an intravenous replacement therapy is offered by Genzyme)
In some cases a diet rich in protein is also advised along with exercise particularly muscle strengthening exercises, but not in patients with cardiac issues.
To date (and to my knowledge), these are the only treatments available. There has been some discussion concerning the use of gene therapy and chaperone therapy to treat Pompe disease. (Gene therapy being the input of the GAA gene into a viral vector and then the infection of a patient's somatic cells. Chaperone therapy being the idea that other small molecules (once transported to the lysosome) can increase the activity of the remaining GAA.)
Citations[]
1. Geel, T.M., McLaughlin, P.M., de Leij, L.F., Ruiters, M.H., and Niezen-Koning, K.E. (2007) Pompe disease: current state of treatment modalities and animal models, Mol Genet Metab 92,299-307.
2. Roach, P.J., Depaoli-Roach, A.A., Hurley, T.D., and Tagliabracci, V.S. Glycogen and it metabolism: some new developments and old themes, Biochem J 441, 763-787.
3. http://ghr.nlm.nih.gov/condition/pompe-disease
4. http://ghr.nlm.nih.gov/gene/GAA
5. Lehninger Principles of Biochemistry. Fifth Edition. Nelson, David, and Cox, Michael. WH Freeman and Company. New York. 6. Kroos, M., Hoogeveen-Westerveld, M., Van Der Ploeg, A., and Reuser, A. (2012) The Genotype-Phenotype Correlation in Pompe Disease. American Journal of Medical Genetics.
7. Banugarie SG, Patel TT, Kishnani PS. Immune modulation in Pompe disease treated with enzyme replacement therapy. Expert Rev Clin Immunol. 2012 Aug: 8(6):497-9.
8. http://www.erasmusmc.nl/klinische_genetica/research/pompe_center/?lang=en
9. Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, Reuser A. The Genotype-Phenotype Correlation in Pompe Disease. American Journal of Medical Genetics. 2012 Jan 17 (online)